62% of patients achieved biochemical response with LIVDELZI1
RESPONSE was a 12-month, randomized, double-blind phase 3 pivotal trial that assessed the efficacy and safety of LIVDELZI 10 mg ± UDCA (n=128) vs placebo ± UDCA (n=65), administered once daily.1
Statistically significant improvement across FDA-accepted markers of cholestasis vs placebo1-3
Composite biochemical response at 12 months1
Primary endpoint1: Composite biochemical response, defined as achieving the following at 12 months:
ALP <1.67 x ULN
≥15% decrease in ALP from baseline
TB ≤1 x ULN
ULN for ALP was defined as 116 U/L for females and males.1,2
Composite biochemical response at 12 months1
Primary endpoint1: Composite biochemical response, defined as achieving the following at 12 months:
ALP <1.67 x ULN
≥15% decrease in ALP from baseline
TB ≤1 x ULN
ULN for ALP was defined as 116 U/L for females and males.1,2
LIVDELZI monotherapy
Biochemical response at 3 months comparing LIVDELZI as a monotherapy with placebo was evaluated in a pooled analysis of a subset of patients from RESPONSE and another randomized, double-blind, placebo-controlled trial in a similar patient population. There was a trend of improvement on biochemical response at 3 months in the LIVDELZI monotherapy group compared with the placebo group.1,a
aIn RESPONSE, 6% of patients in both arms of the trial were UDCA-intolerant and initiated monotherapy (LIVDELZI n=8; placebo n=4).1
LIVDELZIb10 mg oral once daily ± UDCAc (n=128)
Placebobonce daily ± UDCAc (n=65)
Primary endpoint1
Composite biochemical response at 12 months:
ALP <1.67 x ULN
≥15% decrease in ALP from baseline
TB ≤1 x ULN
Key secondary endpoints1
ALP normalization at 12 months, defined as ALP ≤1.0 x ULN
Change from baseline in mean pruritus NRS score at 6 months was analyzed using a mixed-effect model for repeated measuresd
Select secondary endpoint2
Mean change in ALP at 12 months
Inclusion criteria1,2
PBC with an inadequate response or intolerance to UDCA (on treatment ≥12 months; last dose >3 months prior to screening)
ALP ≥1.67 x ULN
TB ≤2 x ULN
ALT/AST ≤3 x ULN
Exclusion criteria1
Other chronic liver diseases
Clinically important hepatic decompensation
Portal hypertension with complications
Cirrhosis with complications (MELD score ≥12, known esophageal varices or variceal bleeds, history of hepatorenal syndrome)
After 12 months, 96% of patients from both arms of the RESPONSE trial opted to enroll in an open-label, long-term study2
bPatients were randomized in a 2:1 ratio.1
cLIVDELZI or placebo was administered in combination with UDCA in 181 (94%) patients during the trial, or as a monotherapy in 12 (6%) patients who were unable to tolerate UDCA.1
dThe pruritus NRS is a scale ranging from 0 (no itch) to 10 (worst itch imaginable).1
LIVDELZI was evaluated in a broad range of patients with PBC2,4
Select baseline demographics and characteristics1,2,4,5
Mean ± SD
Placebo ± UDCA n=65 (%)
LIVDELZI 10 mg ± UDCAn=128 (%)
Demographics
Female, n (%)
60 (92.3)
123 (96.1)
Hispanic/Latino
27 (41.5)
29 (22.7)
Age, years
57.0 ± 9.2
56.6 ± 10.0
Disease characteristics
Duration of disease, years
8.6 ± 6.5
8.2 ± 6.7
Compensated cirrhosis, n (%)
9 (13.8)
18 (14.1)
14% compensated cirrhosis
14% compensated cirrhosis
F2-F3 liver diseasee
17/62 (27.4)
38/115 (33.0)
On UDCA, n (%)
61 (93.8)
120 (93.8)
Pruritus NRS ≥4, n (%)
23 (35.4)
49 (38.3)
Hx of osteopenia, n (%)
8 (12.3)
17 (13.3)
13% or 14% history of osteopenia or osteoporosis
Hx of osteoporosis, n (%)
5 (7.7)
18 (14.1)
13% or 14% history of osteopenia or osteoporosis
Type 2 diabetes
6 (9.2)
15 (11.7)
Liver function values
ALP, U/LULN: 116 U/L
313.8 ± 117.7
314.6 ± 123.0
ALP ≥350 (3 x ULN), n (%)
18 (27.7)
35 (27.3)
ALP <350 U/L, n (%)
47 (72.3)
93 (72.7)
TB, mg/dLULN: 1.1 mg/dL
0.74 ± 0.3
0.77 ± 0.3
TB >1 x ULN & ≤2 x ULN, n (%)
5 (7.7)
20 (15.6)
ALT, U/LULN: 41 U/L
48.2 ± 22.8
47.4 ± 23.5
AST, U/LULN: 34 U/L
41.7 ± 16.0
39.6 ± 16.1
GGT, U/LULN: 52 U/L (M) 38 U/L (F)
287.5 ± 249.6
269.0 ± 240.0
Prior PBC medications
UDCA
65 (100)
128 (100)
OCA use, n (%)
10 (15.4)
15 (11.7)
Fibrates
5 (7.7)
7 (5.5)
Select concomitant use by categoryf,g,h
Lipid-modifying agentsii
27 (41.5)
40 (31.3)
31% taking lipid- modifying agentsi
31% taking lipid-modifying agentsi
Drugs for acid-related disorders
21 (32.3)
47 (36.7)
Sex hormones & modulators
6 (9.2)
7 (5.5)
eThe cutoff values for F1, F2, F3, and F4 were 7.1, 8.8, 10.7, and 16.9 kPa, respectively.5
fSafety analysis set: 128 patients in the LIVDELZI arm and 65 patients in the placebo arm.4,6
gMedications for pruritus were allowed in the study if patients had been on a stable dose for 1 month prior to screening or upon discussion with the medical monitor.4
hReported by Anatomical Therapeutic Classification level 2 terms.4
iBile acid sequestrants: Administer LIVDELZI at least 4 hours before or 4 hours after taking a bile acid sequestrant, or at as great an interval as possible.1
76% biochemical response rate at 12 months observed in a prespecified, exploratory subgroup analysis2,5,6
ALP of 350 U/L is ~3 x ULN1
Biochemical response by ALP level: <350 and ≥350 U/L6
ALP <350 U/L at baseline
76% of patients in the LIVDELZI arm had a biochemical response rate at 12 months vs 23% in the placebo arm (risk difference: 52.9% [95% CI: 36.4, 66.0])5
ALP ≥350 U/L at baseline
23% of patients in the LIVDELZI arm had a biochemical response rate at 12 months vs 11% in the placebo arm (risk difference: 11.7% [95% CI: -12.9, 31.0])5
This analysis is descriptive only and multiplicity was not controlled.2
Biochemical response: ALP <350 U/L6
ALP <350 U/L at baseline
76% of patients in the LIVDELZI arm had a biochemical response rate at 12 months vs 23% in the placebo arm (risk difference: 52.9% [95% CI: 36.4, 66.0])6
This analysis is descriptive only and multiplicity was not controlled.2
Biochemical response: ALP ≥350 U/L6
ALP ≥350 U/L at baseline
23% of patients in the LIVDELZI arm had a biochemical response rate at 12 months vs 11% in the placebo arm (risk difference: 11.7% [95% CI: -12.9, 31.0])6
This analysis is descriptive only and multiplicity was not controlled.2
Important Safety Information
Adverse Reactions
The most common adverse reactions (≥5%) with LIVDELZI were headache (8%), abdominal pain (7%), nausea (6%), abdominal distension (6%), and dizziness (5%).
Please see additional Important Safety Information below.
Observed biochemical response in an open-label, pooled analysis7
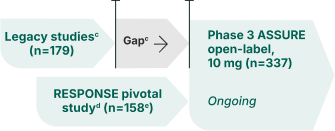
ASSURE is an ongoing, open-label extension (OLE) study evaluating the long-term efficacy and safety results of LIVDELZI. Patients from both arms of the pivotal RESPONSE study or those who were enrolled in a prior LIVDELZI PBC clinical study (legacy studies) were invited to participate in ASSURE. Interim 36-month efficacy and safety results have been reported in a pooled analysis.7,8,a
This is a descriptive analysis and was not designed with statistical control.
Observed biochemical response7
ASSURE is an ongoing, open-label extension (OLE) study evaluating the long-term efficacy and safety results of LIVDELZI. Patients from both arms of the pivotal RESPONSE study or those who were enrolled in a prior LIVDELZI PBC clinical study (legacy studies) were invited to participate in ASSURE. Interim 30-month efficacy and safety results have been reported in a pooled analysis.7,8,a
ASSURE phase 3 open-label study design8,9
Inclusion criteria:
Participated in a PBC study with LIVDELZI
Exclusion criteriab:
AST or ALT >3 x ULN
Total bilirubin >2 x ULN
MELD score ≥12
Evidence of advanced PBC
eGFR ≤45 mL/min/1.73 m2
Use of certain other treatments within 2-3 months prior to screening
History of malignancy diagnosed or treated within 2 years
Use of immunosuppressant therapies
Evidence of advanced PBC
eGFR ≤45 mL/min/1.73 m2
Use of certain other treatments within 2-3 months prior to screening
History of malignancy diagnosed or treated within 2 years
Use of immunosuppressant therapies
Initial data were reported based on exposure to LIVDELZI either upon entry to ASSURE or RESPONSE.
aEfficacy data cutoff of January 31, 2025. Safety data cutoff of January 31, 2024.7,8
bExclusion criteria are only applicable when patients have a study drug interruption of >4 weeks prior to day 1 and for patients who participated in the Phase 1b Hepatic Impairment Study.9
cLegacy seladelpar studies include Phase 2 Dose-Ranging Study (NCT02955602), Phase 3 Long-Term Safety Study (NCT03301506), Phase 3 ENHANCE (NCT03602560), and Phase 1b Hepatic Impairment Study (NCT04950764). Most patients from legacy studies had a treatment gap between their initial use of LIVDELZI and entering the ASSURE study.8,9
dPatients from the placebo arm of the RESPONSE pivotal study switched to active treatment with LIVDELZI in the ASSURE study.9
e54 patients rolled over from the RESPONSE placebo arm and 104 patients rolled over from the RESPONSE LIVDELZI 10 mg arm.8
Important Safety Information
Warnings and Precautions
Fractures: Fractures occurred in 4% of LIVDELZI-treated patients compared to no placebo-treated patients. Consider the risk of fracture in the care of patients treated with LIVDELZI and monitor bone health according to current standards of care.
Please see additional Important Safety Information below.
ALP data
NEW DATA
Open the ASSURE study tab below to see the latest ASSURE ALP data
LIVDELZI is the only PBC treatment that achieved ALP normalization in 25% of patients at 12 months1
RESPONSE was a 12-month, randomized, double-blind phase 3 pivotal trial that assessed the efficacy and safety of LIVDELZI 10 mg ± UDCA (n=128) vs placebo ± UDCA (n=65), administered once daily.1
Help your patients achieve normal ALP with the only PBC treatment to achieve this rate of response1,11
ALP normalization (ALP ≤1.0 x ULN) at 12 months1
Key secondary endpoint1: ALP normalization (ALP ≤1.0 x ULN) at 12 months
In the RESPONSE trial, 6% of patients in both arms of the trial were UDCA-intolerant and initiated monotherapy (LIVDELZI n=8; placebo n=4).2
ALP normalization (ALP ≤1.0 x ULN) at 12 months1
Key secondary endpoint1: ALP normalization (ALP ≤1.0 x ULN) at 12 months
In the RESPONSE trial, 6% of patients in both arms of the trial were UDCA-intolerant and initiated monotherapy (LIVDELZI n=8; placebo n=4).2
Aim for ALP normalization with LIVDELZI1
LIVDELZIa10 mg oral once daily ± UDCAb (n=128)
Placeboaonce daily ± UDCAb (n=65)
Primary endpoint1
Composite biochemical response at 12 months:
ALP <1.67 x ULN
≥15% decrease in ALP from baseline
TB ≤1 x ULN
Key secondary endpoints1
ALP normalization at 12 months, defined as ALP ≤1.0 x ULN
Change from baseline in mean pruritus NRS score at 6 months was analyzed using a mixed-effect model for repeated measuresc
Select secondary endpoint2
Mean change in ALP at 12 months
Inclusion criteria1,2
PBC with an inadequate response or intolerance to UDCA (on treatment ≥12 months; last dose >3 months prior to screening)
ALP ≥1.67 x ULN
TB ≤2 x ULN
ALT/AST ≤3 x ULN
Exclusion criteria1
Other chronic liver diseases
Clinically important hepatic decompensation
Portal hypertension with complications
Cirrhosis with complications (MELD score ≥12, known esophageal varices or variceal bleeds, history of hepatorenal syndrome)
After 12 months, patients from both arms of the RESPONSE trial could enroll in an open-label, long-term study2
aPatients were randomized in a 2:1 ratio.1
bLIVDELZI or placebo was administered in combination with UDCA in 181 (94%) patients during the trial, or as a monotherapy in 12 (6%) patients who were unable to tolerate UDCA.1
cThe pruritus NRS is a scale ranging from 0 (no itch) to 10 (worst itch imaginable).1
LIVDELZI was evaluated in a broad range of patients with PBC2,4
Select baseline demographics and characteristics1,2,4,5
Mean ± SD
Placebo ± UDCA n=65 (%)
LIVDELZI 10 mg ± UDCAn=128 (%)
Demographics
Female, n (%)
60 (92.3)
123 (96.1)
Hispanic/Latino
27 (41.5)
29 (22.7)
Age, years
57.0 ± 9.2
56.6 ± 10.0
Disease characteristics
Duration of disease, years
8.6 ± 6.5
8.2 ± 6.7
Compensated cirrhosis, n (%)
9 (13.8)
18 (14.1)
14% compensated cirrhosis
14% compensated cirrhosis
F2-F3 liver diseased
17/62 (27.4)
38/115 (33.0)
On UDCA, n (%)
61 (93.8)
120 (93.8)
Pruritus NRS ≥4, n (%)
23 (35.4)
49 (38.3)
Hx of osteopenia, n (%)
8 (12.3)
17 (13.3)
13% or 14% history of osteopenia or osteoporosis
Hx of osteoporosis, n (%)
5 (7.7)
18 (14.1)
13% or 14% history of osteopenia or osteoporosis
Type 2 diabetes
6 (9.2)
15 (11.7)
Liver function values
ALP, U/LULN: 116 U/L
313.8 ± 117.7
314.6 ± 123.0
ALP ≥350 (3 x ULN), n (%)
18 (27.7)
35 (27.3)
ALP <350 U/L, n (%)
47 (72.3)
93 (72.7)
TB, mg/dLULN: 1.1 mg/dL
0.74 ± 0.3
0.77 ± 0.3
TB >1 x ULN & ≤2 x ULN, n (%)
5 (7.7)
20 (15.6)
ALT, U/LULN: 41 U/L
48.2 ± 22.8
47.4 ± 23.5
AST, U/LULN: 34 U/L
41.7 ± 16.0
39.6 ± 16.1
GGT, U/LULN: 52 U/L (M) 38 U/L (F)
287.5 ± 249.6
269.0 ± 240.0
Prior PBC medications
UDCA
65 (100)
128 (100)
OCA use, n (%)
10 (15.4)
15 (11.7)
Fibrates
5 (7.7)
7 (5.5)
Select concomitant use by categorye,f,g
Lipid-modifying agentshh
27 (41.5)
40 (31.3)
31% taking lipid- modifying agentsh
31% taking lipid-modifying agentsh
Drugs for acid-related disorders
21 (32.3)
47 (36.7)
Sex hormones & modulators
6 (9.2)
7 (5.5)
dThe cutoff values for F1, F2, F3, and F4 were 7.1, 8.8, 10.7, and 16.9 kPa, respectively.5
eSafety analysis set: 128 patients in the LIVDELZI arm and 65 patients in the placebo arm.4,6
fMedications for pruritus were allowed in the study if patients had been on a stable dose for 1 month prior to screening or upon discussion with the medical monitor.4
gReported by Anatomical Therapeutic Classification level 2 terms.4
hBile acid sequestrants: Administer LIVDELZI at least 4 hours before or 4 hours after taking a bile acid sequestrant, or at as great an interval as possible.1
A rapid and sustained reduction in ALP1
Secondary endpoint2: Mean reduction in ALP over 12 months
ALP change from baseline1,2,12
Mean ALP reduction at months 1, 3, 6, 9, and 12 were secondary endpoints. For these endpoints, multiplicity was not controlled.1,2
At baseline, mean ALP was 314.6 U/L for patients in the LIVDELZI arm and 313.8 U/L for patients in the placebo arm.5
84% of patients
on LIVDELZI had a ≥15% decrease in ALP at 12 months vs 32% of patients in the placebo arm.1
81% of patients in the LIVDELZI arm achieved TB ≤1 x ULN at 12 months vs 77% of patients in the placebo arm.1
In RESPONSE trial, 6% of patients in both arms of the trial were UDCA-intolerant and initiated monotherapy (LIVDELZI n=8; placebo n=4).1
Observed ALP normalization rates in an open-label, pooled analysis7
ASSURE is an ongoing, open-label extension (OLE) study evaluating the long-term efficacy and safety results of LIVDELZI. Patients from both arms of the pivotal RESPONSE study or those who were enrolled in a prior LIVDELZI PBC clinical study (legacy studies) were invited to participate in ASSURE. Interim 36-month efficacy and safety results have been reported in a pooled analysis.7,8,a
This is a descriptive analysis and was not designed with statistical control.
Observed ALP normalization7
ASSURE is an ongoing, open-label extension (OLE) study evaluating the long-term efficacy and safety results of LIVDELZI. Patients from both arms of the pivotal RESPONSE study or those who were enrolled in a prior LIVDELZI PBC clinical study (legacy studies) were invited to participate in ASSURE. Interim 36-month efficacy and safety results have been reported in a pooled analysis.7,8,a
ASSURE phase 3 open-label study design8,9
Inclusion criteria:
Participated in a PBC study with LIVDELZI
Exclusion criteriab:
AST or ALT >3 x ULN
Total bilirubin >2 x ULN
MELD score ≥12
Evidence of advanced PBC
eGFR ≤45 mL/min/1.73 m2
Use of certain other treatments within 2-3 months prior to screening
History of malignancy diagnosed or treated within 2 years
Use of immunosuppressant therapies
Evidence of advanced PBC
eGFR ≤45 mL/min/1.73 m2
Use of certain other treatments within 2-3 months prior to screening
History of malignancy diagnosed or treated within 2 years
Use of immunosuppressant therapies
Initial data were reported based on exposure to LIVDELZI either upon entry to ASSURE or RESPONSE.
aEfficacy data cutoff of January 31, 2025. Safety data cutoff of January 31, 2024.7,8
bExclusion criteria are only applicable when patients have a study drug interruption of >4 weeks prior to day 1 and for patients who participated in the Phase 1b Hepatic Impairment Study.9
cLegacy seladelpar studies include Phase 2 Dose-Ranging Study (NCT02955602), Phase 3 Long-Term Safety Study (NCT03301506), Phase 3 ENHANCE (NCT03602560), and Phase 1b Hepatic Impairment Study (NCT04950764). Most patients from legacy studies had a treatment gap between their initial use of LIVDELZI and entering the ASSURE study.8,9
dPatients from the placebo arm of the RESPONSE pivotal study switched to active treatment with LIVDELZI in the ASSURE study.9
e54 patients rolled over from the RESPONSE placebo arm and 104 patients rolled over from the RESPONSE LIVDELZI 10 mg arm.8
Post-hoc subgroup analysis of composite ALP normalization in patients with ALP of 1.0-1.67 x ULN at baseline entry into ASSURE10,f
This exploratory post-hoc analysis is descriptive only and was not designed with statistical control and statistical analyses were not performed. This data was not type 1 error or multiplicity controlled.
Composite ALP normalization was defined as achieving both10:
ALP ≤1.0 x ULN (116 U/L for females and males2)
≥15% ALP reduction
Observed composite ALP normalization10,f
Upon entry into the open-label ASSURE study, the subgroup, pooled analysis included 50 patients from the LIVDELZI legacy studies.10
At baseline, mean ALP was 156 U/L.10
fPatients in phase 2 and 3 LIVDELZI legacy studies had a required baseline ALP ≥1.67 x ULN.10
Person featured is compensated by Gilead.
“Now that I’m taking LIVDELZI, I’m looking forward to seeing what this PBC treatment can do.”
Jennifer
Real LIVDELZI patient
Person featured is compensated by Gilead.
"After being on LIVDELZI about a month, my ALP was already moving toward normal."
Wendy
Real LIVDELZI patient
Person featured is compensated by Gilead.
"After starting LIVDELZI, I got my labs and my ALP was the lowest it had been since I was diagnosed."
Amanda
Real LIVDELZI patient
Pruritus data
For patients with baseline average pruritus NRS score ≥41
LIVDELZI is the only PBC treatment with statistically significant pruritus improvement at 6 months in the FDA-approved label1
RESPONSE was a 12-month, randomized, double-blind phase 3 pivotal trial that assessed the efficacy and safety of LIVDELZI 10 mg ± UDCA (n=128) vs placebo ± UDCA (n=65), administered once daily.1
Key secondary endpoint1: Change in pruritus NRS at 6 months in patients with baseline average pruritus score ≥4
Reduction in pruritus with LIVDELZI was1,2:
A single-item, patient-reported outcome, the pruritus NRS, evaluated patients’ daily worst itching intensity on an 11-point rating scale, with scores ranging from 0 (“no itching”) to 10 (“worst itching imaginable”) in RESPONSE. The pruritus NRS was administered daily in a 14-day run-in period prior to randomization through 6 months and then for 7 consecutive days during each month up to the end of the treatment period.1,2
Change from baseline in pruritus NRS in patients with baseline average pruritus score ≥41,2
Pruritus NRS is a validated itch scale recognized by the FDA to measure the statistical significance or pruritus improvement in clinical trials13
Pruritus NRS scores at 1 and 9 months were exploratory endpoints and at 3 and 12 months were secondary endpoints. For these endpoints, multiplicity was not controlled.2
The baseline average pruritus score for each patient was calculated by averaging the pruritus NRS scores administered in the run-in period and on day 1 before treatment initiation. The pruritus scores at 6 months for each patient were calculated by averaging the pruritus NRS scores within the last week in the month.1
LIVDELZIa10 mg oral once daily ± UDCAb (n=128)
Placeboaonce daily ± UDCAb (n=65)
Primary endpoint1
Composite biochemical response at 12 months:
ALP <1.67 x ULN
≥15% decrease in ALP from baseline
TB ≤1 x ULN
Key secondary endpoints1
ALP normalization at 12 months, defined as ALP ≤1.0 x ULN
Change from baseline in mean pruritus NRS score at 6 months was analyzed using a mixed-effect model for repeated measuresc
Select secondary endpoint2
Mean change in ALP at 12 months
Inclusion criteria1,2
PBC with an inadequate response or intolerance to UDCA (on treatment ≥12 months; last dose >3 months prior to screening)
ALP ≥1.67 x ULN
TB ≤2 x ULN
ALT/AST ≤3 x ULN
Exclusion criteria1
Other chronic liver diseases
Clinically important hepatic decompensation
Portal hypertension with complications
Cirrhosis with complications (MELD score ≥12, known esophageal varices or variceal bleeds, history of hepatorenal syndrome)
After 12 months, patients from both arms of the RESPONSE trial could enroll in an open-label, long-term study2
aPatients were randomized in a 2:1 ratio.1
bLIVDELZI or placebo was administered in combination with UDCA in 181 (94%) patients during the trial, or as a monotherapy in 12 (6%) patients who were unable to tolerate UDCA.1
cThe pruritus NRS is a scale ranging from 0 (no itch) to 10 (worst itch imaginable).1
LIVDELZI was evaluated in a broad range of patients with PBC2,4
Select baseline demographics and characteristics1,2,4,5
Mean ± SD
Placebo ± UDCA n=65 (%)
LIVDELZI 10 mg ± UDCAn=128 (%)
Demographics
Female, n (%)
60 (92.3)
123 (96.1)
Hispanic/Latino
27 (41.5)
29 (22.7)
Age, years
57.0 ± 9.2
56.6 ± 10.0
Disease characteristics
Duration of disease, years
8.6 ± 6.5
8.2 ± 6.7
Compensated cirrhosis, n (%)
9 (13.8)
18 (14.1)
14% compensated cirrhosis
14% compensated cirrhosis
F2-F3 liver diseased
17/62 (27.4)
38/115 (33.0)
On UDCA, n (%)
61 (93.8)
120 (93.8)
Pruritus NRS ≥4, n (%)
23 (35.4)
49 (38.3)
Hx of osteopenia, n (%)
8 (12.3)
17 (13.3)
13% or 14% history of osteopenia or osteoporosis
Hx of osteoporosis, n (%)
5 (7.7)
18 (14.1)
13% or 14% history of osteopenia or osteoporosis
Type 2 diabetes
6 (9.2)
15 (11.7)
Liver function values
ALP, U/LULN: 116 U/L
313.8 ± 117.7
314.6 ± 123.0
ALP ≥350 (3 x ULN), n (%)
18 (27.7)
35 (27.3)
ALP <350 U/L, n (%)
47 (72.3)
93 (72.7)
TB, mg/dLULN: 1.1 mg/dL
0.74 ± 0.3
0.77 ± 0.3
TB >1 x ULN & ≤2 x ULN, n (%)
5 (7.7)
20 (15.6)
ALT, U/LULN: 41 U/L
48.2 ± 22.8
47.4 ± 23.5
AST, U/LULN: 34 U/L
41.7 ± 16.0
39.6 ± 16.1
GGT, U/LULN: 52 U/L (M) 38 U/L (F)
287.5 ± 249.6
269.0 ± 240.0
Prior PBC medications
UDCA
65 (100)
128 (100)
OCA use, n (%)
10 (15.4)
15 (11.7)
Fibrates
5 (7.7)
7 (5.5)
Select concomitant use by categorye,f,g
Lipid-modifying agentshh
27 (41.5)
40 (31.3)
31% taking lipid- modifying agentsh
31% taking lipid-modifying agentsh
Drugs for acid- related disorders
21 (32.3)
47 (36.7)
Sex hormones & modulators
6 (9.2)
7 (5.5)
dThe cutoff values for F1, F2, F3, and F4 were 7.1, 8.8, 10.7, and 16.9 kPa, respectively.5
eSafety analysis set: 128 patients in the LIVDELZI arm and 65 patients in the placebo arm.4,6
fMedications for pruritus were allowed in the study if patients had been on a stable dose for 1 month prior to screening or upon discussion with the medical monitor.4
gReported by Anatomical Therapeutic Classification level 2 terms.4
hBile acid sequestrants: Administer LIVDELZI at least 4 hours before or 4 hours after taking a bile acid sequestrant, or at as great an interval as possible.1
Person featured is compensated by Gilead.
"Before LIVDELZI, I felt like I could never scratch deep enough and could never hit the spot. But now my itching has improved."
Amanda
Real LIVDELZI patient
Person featured is compensated by Gilead.
"I was bleeding and covered in sores all over my body from scratching. After starting LIVDELZI my itch improved a lot."
Christine
Real LIVDELZI patient
Person featured is compensated by Gilead.
"With LIVDELZI my itching improved, and I'm not itching as much throughout the night.”
Wendy
Real LIVDELZI patient
Exploratory PBC-40 fatigue & pruritus data
Exploratory PBC-40 patient-reported outcomes on fatigue and pruritus4,5
Fatigue and pruritus were evaluated in prespecified, exploratory analyses2,4,5
These analyses are descriptive only and multiplicity was not controlled.4
OVERALL INTENT-TO-TREAT POPULATION
PBC-40 domain scores: mean change from baseline at 12 months
Fatigue14
Baseline average fatigue scores: 27.6 (LIVDELZI), 27.4 (placebo).14
Pruritus4,5
Baseline average itch scores: 5.1 (LIVDELZI), 5.6 (placebo).14
PBC-40
A validated health-related questionnaire designed to quantify the impact of PBC across 6 domains15
In the PBC-40, patients respond to 40 PBC-specific impact questions in which higher scores represent greater impact. Domains include2:
Fatigue
Social
Symptoms
Cognition
Emotion
Itch
PBC-40 domains of fatigue and pruritus were exploratory endpoints. For these endpoints, multiplicity was not controlled. Analyses are not presented in the full Prescribing Information for LIVDELZI.2,4
The PBC-40 Fatigue domain ranges from 11 to 55 (lowest to highest impact). The PBC-40 Itch domain ranges from 3 to 15 (lowest to highest impact).15
LIVDELZIa10 mg oral once daily ± UDCAb (n=128)
Placeboaonce daily ± UDCAb (n=65)
Primary endpoint1
Composite biochemical response at 12 months:
ALP <1.67 x ULN
≥15% decrease in ALP from baseline
TB ≤1 x ULN
Key secondary endpoints1
ALP normalization at 12 months, defined as ALP ≤1.0 x ULN
Change from baseline in mean pruritus NRS score at 6 months was analyzed using a mixed-effect model for repeated measuresc
Select secondary endpoint2
Mean change in ALP at 12 months
Inclusion criteria1,2
PBC with an inadequate response or intolerance to UDCA (on treatment ≥12 months; last dose >3 months prior to screening)
ALP ≥1.67 x ULN
TB ≤2 x ULN
ALT/AST ≤3 x ULN
Exclusion criteria1
Other chronic liver diseases
Clinically important hepatic decompensation
Portal hypertension with complications
Cirrhosis with complications (MELD score ≥12, known esophageal varices or variceal bleeds, history of hepatorenal syndrome)
After 12 months, patients from both arms of the RESPONSE trial could enroll in an open-label, long-term study2
aPatients were randomized in a 2:1 ratio.1
bLIVDELZI or placebo was administered in combination with UDCA in 181 (94%) patients during the trial, or as a monotherapy in 12 (6%) patients who were unable to tolerate UDCA.1
cThe pruritus NRS is a scale ranging from 0 (no itch) to 10 (worst itch imaginable).1
LIVDELZI was evaluated in a broad range of patients with PBC2,4
Select baseline demographics and characteristics1,2,4,5
Mean ± SD
Placebo
± UDCA n=65 (%)
LIVDELZI
10 mg ± UDCAn=128 (%)
Demographics
Female, n (%)
60 (92.3)
123 (96.1)
Hispanic/Latino
27 (41.5)
29 (22.7)
Age, years
57.0 ± 9.2
56.6 ± 10.0
Disease characteristics
Duration of disease, years
8.6 ± 6.5
8.2 ± 6.7
Compensated cirrhosis, n (%)
9 (13.8)
18 (14.1)
14% compensated
cirrhosis
14% compensated cirrhosis
F2-F3 liver diseased
17/62 (27.4)
38/115 (33.0)
On UDCA, n (%)
61 (93.8)
120 (93.8)
Pruritus NRS ≥4, n (%)
23 (35.4)
49 (38.3)
Hx of osteopenia, n (%)
8 (12.3)
17 (13.3)
13% or 14% history of
osteopenia or
osteoporosis
Hx of osteoporosis, n (%)
5 (7.7)
18 (14.1)
13% or 14% history of osteopenia or osteoporosis
Type 2 diabetes
6 (9.2)
15 (11.7)
Liver function values
ALP, U/LULN:
116 U/L
313.8 ± 117.7
314.6 ± 123.0
ALP ≥350
(3 x ULN), n (%)
18 (27.7)
35 (27.3)
ALP <350 U/L, n (%)
47 (72.3)
93 (72.7)
TB, mg/dLULN:
1.1 mg/dL
0.74 ± 0.3
0.77 ± 0.3
TB >1 x ULN & ≤2 x ULN, n (%)
5 (7.7)
20 (15.6)
ALT, U/LULN:
41 U/L
48.2 ± 22.8
47.4 ± 23.5
AST, U/LULN:
34 U/L
41.7 ± 16.0
39.6 ± 16.1
GGT, U/LULN:
52 U/L (M)
38 U/L (F)
287.5 ± 249.6
269.0 ± 240.0
Prior PBC medications
UDCA
65 (100)
128 (100)
OCA use, n (%)
10 (15.4)
15 (11.7)
Fibrates
5 (7.7)
7 (5.5)
Select concomitant use by categorye,f,g
Lipid-modifying
agentshh
27 (41.5)
40 (31.3)
31% taking lipid-
modifying agentsh
31% taking lipid-modifying agentsh
Drugs for acid- related disorders
21 (32.3)
47 (36.7)
Sex hormones &
modulators
6 (9.2)
7 (5.5)
dThe cutoff values for F1, F2, F3, and F4 were 7.1, 8.8, 10.7, and 16.9 kPa, respectively.5
eSafety analysis set: 128 patients in the LIVDELZI arm and 65 patients in the placebo arm.4,6
fMedications for pruritus were allowed in the study if patients had been on a stable dose for 1 month prior to screening or upon discussion with the medical monitor.4
gReported by Anatomical Therapeutic Classification level 2 terms.4
hBile acid sequestrants: Administer LIVDELZI at least 4 hours before or 4 hours after taking a bile acid sequestrant, or at as great an interval as possible.1
A post-hoc subgroup analysis of PBC-40 fatigue and pruritus outcomes based on baseline NRS pruritus scores2,4
This post-hoc analysis was not designed with statistical control and statistical comparisons were not performed. These data were not type 1 error controlled. These exploratory analyses are descriptive only and multiplicity was not controlled.2,5
SUBGROUP ANALYSIS: NRS ≥7 at baseline
PBC-40 mean scores through 12 months14
Fatigue
Baseline average fatigue scores: 31.3 (LIVDELZI) and 36.3 (placebo).14
Pruritus
Baseline average itch scores: 10.6 (LIVDELZI) and 10.5 (placebo).14
SUBGROUP ANALYSIS: NRS ≥4 at baseline
PBC-40 mean scores through 12 months14
Fatigue
Baseline average fatigue scores: 31.9 (LIVDELZI) and 34.7 (placebo).14
Pruritus4,14
Baseline average itch scores: 8.7 (LIVDELZI) and 9.6 (placebo).14
PBC-40 analyses are not presented in the LIVDELZI full Prescribing Information.
Review the safety and tolerability profile of LIVDELZI
LIVDELZI is indicated for the treatment of primary biliary cholangitis (PBC), in combination with ursodeoxycholic acid (UDCA) in adults who have had an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA.
This indication is approved under accelerated approval based on a reduction of alkaline phosphatase (ALP). Improvement in survival or prevention of liver decompensation events have not been demonstrated. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s).
Limitation of Use: Use of LIVDELZI is not recommended in patients who have or develop decompensated cirrhosis (eg, ascites, variceal bleeding, hepatic encephalopathy).
IMPORTANT SAFETY INFORMATION
Warnings and Precautions
Fractures: Fractures occurred in 4% of LIVDELZI-treated patients compared to no placebo-treated patients. Consider the risk of fracture in the care of patients treated with LIVDELZI and monitor bone health according to current standards of care.
Liver Test Abnormalities: LIVDELZI has been associated with dose-related increases in serum transaminase (AST and ALT) levels >3x ULN in patients receiving 50 mg and 200 mg once daily (5x and 20x higher than the recommended dosage of 10 mg once daily). Perform baseline clinical and laboratory testing when starting LIVDELZI and monitor thereafter according to routine patient management. Interrupt treatment if the liver tests (ALT, AST, total bilirubin, and/or ALP) worsen, or if the patient develops signs and symptoms of clinical hepatitis (eg, jaundice, right upper quadrant pain, eosinophilia). Consider permanent discontinuation if liver tests worsen after restarting LIVDELZI.
Biliary Obstruction: Avoid use of LIVDELZI in patients with complete biliary obstruction. If biliary obstruction is suspected, interrupt LIVDELZI and treat as clinically indicated.
Adverse Reactions
The most common adverse reactions (≥5%) with LIVDELZI were headache (8%), abdominal pain (7%), nausea (6%), abdominal distension (6%), and dizziness (5%).
Drug Interactions
Potential Increased Exposure of LIVDELZI with:
Probenecid: Avoid coadministration with LIVDELZI.
Strong CYP2C9 Inhibitors: Monitor for adverse effects during concomitant use.
Dual Moderate CYP2C9 and Moderate or Strong CYP3A4 Inhibitors (eg, fluconazole): Monitor for adverse effects during concomitant use.
CYP2C9 Poor Metabolizers Using Moderate or Strong CYP3A4 Inhibitors: Monitor for adverse effects during concomitant use of a moderate or strong CYP3A4 inhibitor in patients who are CYP2C9 poor metabolizers.
Dual or Multiple Clinical Inhibitors of Drug Transporters OATP1B1, OATP1B3, and BCRP (eg, cyclosporine): Monitor for adverse effects during concomitant use.
Potential Reduction in LIVDELZI Exposure with:
Rifampin: Concomitant use of LIVDELZI with rifampin, an inducer of metabolizing enzymes, may result in delayed or suboptimal LIVDELZI biochemical response. Monitor biochemical response (eg, ALP and bilirubin) when patients initiate rifampin during LIVDELZI treatment.
Bile Acid Sequestrants: Administer LIVDELZI at least 4 hours before or 4 hours after taking a bile acid sequestrant, or at as great an interval as possible.
Pregnancy and Lactation
Pregnancy: There are insufficient data from human pregnancies exposed to LIVDELZI to allow an assessment of a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Report pregnancies to Gilead Sciences, Inc., at 1-800-445-3235.
Lactation: There are no data on the presence of LIVDELZI in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for LIVDELZI and any potential adverse effects on the breastfed infant from LIVDELZI.
LIVDELZI [prescribing information]. Foster City, CA: Gilead Sciences, Inc.; 2026.
Hirschfield GM, Bowlus CL, Mayo MJ, et al; RESPONSE Study Group. A phase 3 trial of seladelpar in primary biliary cholangitis. N Engl J Med. 2024;390(9):783-794. doi:10.1056/NEJMoa2312100
Lammers WJ, van Buuren HR, Hirschfield GM, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology. 2014;147(6):1338-1349.e5. doi:10.1053/j.gastro.2014.08.029
Hirschfield GM, Bowlus CL, Mayo MJ, et al; RESPONSE Study Group. A phase 3 trial of seladelpar in primary biliary cholangitis [supplementary appendix]. N Engl J Med. 2024;390(9):783-794. doi:10.1056/NEJMoa2312100
Data on file. Gilead Sciences, Inc.; 2024.
Data on file [supplementary appendix]. Gilead Sciences, Inc.; 2024.
Pratt D, Lawitz EJ, Bowlus CL, et al. Seladelpar leads to sustained reduction in cholestatic markers with consistent safety profile in patients with primary biliary cholangitis up to 48 months. Poster presented at: The Liver Meeting, American Association for the Study of Liver Diseases; November 7-11, 2025; Washington, DC.
Lawitz EJ, Trivedi PJ, Kowdley KV, et al. Long-term efficacy and safety of open-label seladelpar treatment in patients with primary biliary cholangitis: pooled interim results for up to 3 years from the ASSURE study. Poster presented at: The Liver Meeting, American Association for the Study of Liver Diseases; November 15-19, 2024; San Diego, CA.
Yimam K, Levy C, Kowdley KV, et al. ASSURE study in progress: an open-label long-term extension study to evaluate the safety and tolerability of seladelpar in subjects with primary biliary cholangitis (PBC). Poster presented at: Liver Connect Conference; April 4-6, 2024; Scottsdale, AZ.
Data on file. Gilead Sciences, Inc.; 2026.
Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69(1):394-419. doi:10.1002/hep.30145
Data on file. Gilead Sciences, Inc.; 2024.
Yosipovitch G, Reaney M, Mastey V, et al. Peak Pruritus Numerical Rating Scale: psychometric validation and responder definition for assessing itch in moderate-to-severe atopic dermatitis. Br J Dermatol. 2019;181(4):761-769. doi:10.1111/bjd.17744
Data on file. Gilead Sciences, Inc.; 2024.
Jacoby A, Rannard A, Buck D, et al. Development, validation, and evaluation of the PBC-40, a disease specific health related quality of life measure for primary biliary cirrhosis. Gut. 2005;54(11):1622-1629. doi:10.1136/gut.2005.065862
This information is intended for US healthcare professionals
By selecting “Continue” below, you verify that you are a US healthcare professional.